Introducción
Los fármacos antinflamatorios no esteroideos (AINE) son utilizados frecuentemente en la práctica clínica para el tratamiento del dolor y/o la inflamación. Los AINE son un grupo de fármacos heterogéneo, con un mecanismo de acción común (inhibición de las enzimas COX) pero con diferentes perfiles farmacodinámicos y farmacocinéticos, resultando en unos perfiles de eficacia, seguridad e interacciones diferentes de cada molécula. Es importante mantenerse actualizado sobre estas diferencias para poder elegir adecuadamente el fármaco óptimo en cada situación. El objetivo de esta revisión es ofrecer una actualización de todo lo que se conoce sobre los AINE, destacando las diferencias y peculiaridades de cada molécula.
Para abordar esta revisión nos parece relevante abordar los siguientes apartados:
— Orígenes históricos de los AINE.
— Definición y clasificación.
— Mecanismo de acción de cada molécula.
— Efectos terapéuticos.
— Farmacocinética.
— Interacciones más relevantes.
— Efectos adversos más importantes a tener en cuenta en la práctica clínica habitual.
El objetivo de este trabajo es ofrecer a los profesionales sanitarios un documento único de repaso sobre los puntos más relevantes de este grupo de fármacos.
Orígenes históricos de los AINE: la aspirina y el ibuprofeno
La salicilina, un glucósido contenido en la corteza de los sauces pertenecientes a la familia de las salicáceas (género salix, como salix alba, salix fragilis) es probablemente el remedio más antiguo contra el dolor que se usa actualmente (1). Los antiguos egipcios usaban extractos de mirto y hojas de sauce para calmar los dolores en las articulaciones. De hecho, el papiro Ebers (1534 a. C.) informa sobre aplicaciones de decocciones de hojas de mirto y sauce en el abdomen y la espalda para aliviar condiciones inflamatorias y síntomas dolorosos (2). Unos 1000 años más tarde, Hipócrates de Kos (460-377 a. C.), el padre de la medicina, y luego Galeno de Pérgamo (129-201 d. C.), eran conscientes de las propiedades medicinales de las plantas de Salicaceae. Hipócrates prescribió corteza de sauce para tratar el dolor inflamatorio y aliviar el dolor del parto (3).
En 1758, el reverendo Edward Stone Oxfordshire de Inglaterra, estudió las propiedades curativas del sauce, buscando un remedio válido y más barato que la costosa corteza de quina para tratar los síntomas de la malaria (4). Administró extracto acuoso de corteza de Salix alba a 50 pacientes con fiebre, y descubrió que la administración de estos extractos cada 4 h tenía una marcada acción antipirética. Stone presentó este estudio a la Royal Society de Londres en 1763 (5), y es reconocido como el primer autor en demostrar, con rigor científico, la eficacia de la corteza de sauce en el tratamiento de la fiebre.
La primera extracción del componente activo de la corteza de sauce fue realizada en 1824 por dos farmacéuticos italianos de Verona, Francesco Fontana y Bartolommeo Rigatelli (6). Joseph Buchner, un farmacólogo alemán, también extrajo el ingrediente activo del sauce, produciendo cristales amarillos de sabor amargo, que llamó “salicina” (7). En 1829, el farmacéutico francés Henri Leroux perfeccionó el proceso de extracción de salicina y aisló cantidades importantes de cristales solubles de salicina pura (8).
En 1838, Raffaele Pirìa, uno de los químicos italianos más importantes del siglo xix, extrajo ácido salicílico de la salicina separando el componente de glucosa, y determinó su fórmula molecular (9). En los años siguientes, la investigación se dirigió hacia la síntesis química del ácido salicílico para obtener un compuesto más puro, mejor tolerado y menos costoso. En 1853, el químico francés Charles Gerhardt demostró por primera vez la reacción del salicilato de sodio con cloruro de acetilo, que daba lugar a una sustancia blanca que llamó “acide acéto-salicylique”. De hecho, obtuvo ácido acetilsalicílico (AAS), aspirina, sin saberlo. El compuesto clave para la síntesis de la aspirina, el ácido salicílico, fue descrito químicamente y sintetizado en 1859 por Hermann Kolbe, profesor de química en la Universidad de Marburgo, Alemania (3). En 1874, estableció la primera fábrica para la producción de salicilatos sintéticos en Dresde, y Von Heyden fue el primero en recibir una patente realizando la comercialización del compuesto a un precio diez veces más bajo que los extractos de sauce (10).
El desarrollo de salicilatos se reanudó en los laboratorios de Bayer, fundados en 1863 por Friedrich Bayer y William Weskott, inicialmente especializados en la producción de tintes. En 1888, la compañía decidió establecer una división de medicamentos encabezada por Carl Duisberg, con una rama farmacéutica encabezada por Arthur Eichengrün y una rama farmacológica, encabezada por Heinrich Dreser. Querían sintetizar un derivado de los salicilatos que no causara los efectos adversos (principalmente náuseas, irritación gástrica y tinnitus) comunes con el uso de salicilato de sodio. Esta tarea fue asignada a Felix Hoffmann, un joven químico farmacéutico que se graduó en la Universidad de Munich (3,11). Hoffmann era químico en Bayer; pasó a acetilar el grupo fenol del ácido salicílico produciendo un ácido acetilsalicílico estable puro (ASA) (12) el 10 de agosto de 1897, según su cuaderno de laboratorio, utilizando una reacción de acetilación (acetilar el grupo fenol a partir de ácido salicílico refluido con anhídrido acético), obteniendo ácido acetilsalicílico en su forma más pura con un método relativamente simple, fiable y eficiente. Esa fecha (10 de agosto de 1897) ha sido acreditada desde entonces como el nacimiento de la aspirina. El nuevo medicamento recibió el nombre de “aspirina” y el 1 de febrero de 1899, Bayer registró el nombre de la marca en Berlín, siendo patentada en los Estados Unidos en 1900 (3).
Por tanto, la primera generación de AINE se comenzó a comercializar en 1899 gracias al farmacólogo alemán Félix Hoffman con el nombre de Aspirina®, término acuñado por los laboratorios Bayer (se le dio el nombre tomando la A [de acetilación] y la palabra griega spiraea, del nombre científico de la planta ulmaria [Spiraea ulmaria] donde se obtiene el AAS) (13).
Inicialmente comercializado en forma de polvo, el medicamento se puso a disposición en 1904 en forma de tabletas, y como tal fue el primer medicamento industrial disponible en esta forma farmacéutica en todo el mundo (14), teniendo un éxito comercial inmediato.
Los primeros estudios clínicos sobre la aspirina fueron publicados en 1899, en Alemania, por Witthauer (15) y Wohlgemut (16), y en los Estados Unidos por Floeckinger (17).
La disponibilidad de la aspirina como fármaco antinflamatorio modelo (con un perfil muy característico de efectos biológicos) condujo al desarrollo de otros fármacos con acciones similares: la fenilbutazona se introdujo en la década de 1940, los fenamatos en la década de 1950, la indometacina en la década de 1960, los propionatos en la década de 1970 y los oxicams en la década de 1980 (18).
La aspirina fue la primera molécula de un conjunto de fármacos que se llamarían posteriormente antinflamatorios no esteroideos (AINE), un grupo con una farmacología ampliamente similar a la de la aspirina pero estructuralmente distintos y con perfiles diferentes, aunque la mayoría son ácidos carboxílicos (19).
Antes de 1970 se sabía poco sobre el mecanismo de acción de la aspirina. John Vane y sus cols. descubrieron el mecanismo de acción de la aspirina y otros AINE (20), allanando así el camino para el desarrollo de nuevos medicamentos antinflamatorios (21). Vane descubrió que la aspirina bloqueaba la acción de la enzima ciclooxigenasa (COX-1), una enzima que induce la formación de prostaglandinas (20). Estos lípidos se habían asociado a la producción de dolor, fiebre e inflamación, así como en la prevención del daño gástrico, por lo que este mecanismo proporcionó una explicación completa para las conocidas acciones analgésicas, antinflamatorias, antipiréticas y gastrotóxicas de la aspirina. Vane y sus cols. demostraron que la aspirina, la indometacina y el salicilato de sodio bloqueaban la síntesis de prostaglandinas (PG) en sistemas libres de células y en el bazo de un perro perfundido aislado ex vivo. Un estudio adicional en voluntarios humanos que tomaron una dosis terapéutica de aspirina mostró que los niveles de PG se redujeron en muestras de líquido seminal y agregación de plaquetas, confirmando que los AINE pueden inhibir la síntesis de PG al dirigirse directamente a la enzima COX, tanto in vitro como in vivo (22).
El siguiente gran avance en el campo de los AINE ocurrió a principios de la década de 1990, cuando se descubrió una isoforma inducible de COX, denominada COX-2 (23). Se propuso que esta era la isoforma más significativa de la enzima COX en la inflamación, pero menos importante como enzima gastroprotectora en el estómago. Se encontró que los AINE convencionales de la época eran inhibidores no selectivos que actuaban sobre la COX-1 y la COX-2 en diversos grados, tal vez explicando por qué tenían efectos terapéuticos y tóxicos mixtos. Esto condujo, a su vez, al desarrollo de un nuevo concepto: que los inhibidores selectivos de la COX-2 deberían tener efectos antinflamatorios superiores, pero menos adversos gastrointestinales que los AINE tradicionales (no selectivos) (24).
El descubrimiento posterior de los “coxibs”, inhibidores más selectivos de la COX-2, como celecoxib, rofecoxib, lumiracoxib y valdecoxib, proporcionó opciones adicionales para tratar a los pacientes que sufrían de irritación gástrica, aunque, decepcionantemente no eliminaron el problema por completo.
Tanto los AINE originales como los coxibs más nuevos siguen siendo el pilar de la terapia farmacológica antinflamatoria para el dolor y la hinchazón en la osteoartritis y la artritis reumatoide, afecciones inflamatorias agudas (como lesiones deportivas, fracturas y lesiones de tejidos blandos) y postoperatorio, dolor de cabeza, migraña dental, dolor menstrual y algunas condiciones de trauma agudo (25).
Cincuenta años después de conocerse el mecanismo de acción de la aspirina, el nuevo milenio había traído consigo una reevaluación de nuestra comprensión de los efectos terapéuticos y secundarios de estos medicamentos; las explicaciones de la cardiotoxicidad de los AINE y la toxicidad gastrointestinal siguen siendo temas controvertidos. Aunque el mecanismo exacto de acción sobre cómo estos medicamentos se dirigen a las células cancerosas no está bien dilucidado, los estudios han demostrado que la aspirina previene el cáncer colorrectal a través de la inhibición de NF-κB y las vías de señalización (26). Se han propuesto nuevas dianas de la aspirina y el ácido salicílico, como la ciclina A2 y CDK2, entre otras, como mecanismo de acción preventiva contra el cáncer (27).
Por el contrario, se ha demostrado que la aspirina acetila la isoforma inflamatoria COX-2, lo que lleva a la producción de mediadores lipídicos prosolutivos, derivados de omega-3 (28). Curiosamente, la aspirina se había asociado con una reducción más lenta de la capacidad cognitiva, particularmente en la enfermedad de Alzheimer (29), al dirigirse al factor de transcripción nuclear proliferador de peroxisomas gamma (PPAR-γ) (30) y a través de la regulación a la baja de COX-1 y COX-2 (31).
La clasificación de los medicamentos AINE generalmente se basa en la estructura química y la selectividad de la COX: los inhibidores no selectivos incluyen salicilatos acetilados (aspirina), salicilatos no acetilados (diflunisal), ácidos acéticos (indometacina, diclofenaco), ácidos propiónicos (ibuprofeno, naproxeno), ácidos enólicos (piroxicam, meloxicam) y los inhibidores más selectivos de la COX-2 (celecoxib, etoricoxib) (18).
Muchos AINE están disponibles en una variedad de formulaciones, como tabletas, inyecciones y geles, y varios están disponibles en farmacias sin receta.
A pesar de que el concepto de Vane había llevado a una detección in vitro simple pero robusta de supuestos compuestos antinflamatorios que inhiben una enzima COX (32), se observaron varias anomalías. Por ejemplo, el paracetamol (acetaminofeno) es un medicamento muy prominente que se introdujo a fines de 1800 como un medicamento antipirético y también posee efectos analgésicos, como otros medicamentos similares a la aspirina (33,34). Sin embargo, a diferencia de este último, el paracetamol tiene muy poca actividad antinflamatoria y está completamente desprovisto de efectos gástricos o plaquetarios (35). De acuerdo con su perfil terapéutico, se encontró que el paracetamol era más efectivo contra las preparaciones de COX cerebral crudas en comparación con las aisladas de tejidos periféricos, como el bazo, lo que sugiere una explicación putativa para la selectividad de su acción terapéutica y la noción de que había varias formas de la enzima que se formularon (34). En animales de experimentación, la analgesia y la antipiresis con paracetamol se acompañan de la reducción de la síntesis de prostaglandinas en el sistema nervioso central (34,36).
Como el paracetamol es solo un inhibidor débil de las actividades de la COX-1 y la COX-2, sus acciones farmacológicas no podrían explicarse por la inhibición de estas enzimas (34). Curiosamente, la COX-3, que se identificó como una variante de empalme de la COX-1 en 2002 en tejidos caninos, se demostró ser inhibida por el paracetamol. Por lo tanto, se esperaba que el descubrimiento de la COX-3 pudiera proporcionar una explicación clara de las acciones farmacológicas del paracetamol (37).
Ibuprofeno (38)
Stewart Sanders Adams es considerado el padre del ibuprofeno. Fue líder de un equipo de investigación en Boots, Inglaterra. Su primer trabajo fue en la producción de penicilina en los primeros años de su desarrollo. Como investigador, su primer trabajo en 1948 fue con heparina, intentando posteriormente desarrollar un fármaco heparinoide con características antilipídicas y poco anticoagulante. Sus estudios con la heparina le llevaron a creer que la heparina y la histamina co-existían en las células mastocito. En 1952 empezó a trabajar en artritis reumatoide, ya que en esos años las únicas sustancias con valor terapéutico para esta indicación eran los corticoides y dosis altas de aspirina. La necesidad de obtener una sustancia sin efectos adversos gastrointestinales y sin los efectos de los corticoides, impulsó su investigación. Los efectos antinflamatorios de la aspirina todavía no eran conocidos, y Adams creyó que sus efectos analgésicos tenían que deberse a un efecto antinflamatorio, que intentó estudiar en modelos animales, demostrando que la diferencia entre la aspirina y el paracetamol era la capacidad del primero de producir antinflamación.
La aspirina se estaba usando desde 1900 y todavía no se había desarrollado ningún análogo, por lo que Adams se unió a Nicholson, un químico que había testado unos 200 análogos sin demostrar superioridad a la aspirina. Con modificaciones adicionales, el químico Nicholson dio lugar a unos nuevos compuestos (ácidos fenilacéticos), tres de los cuales eran activos en artritis reumatoide. Los estudios dieron después lugar a la identificación de ácidos fenil-propiónicos.
Durante ese tiempo, cuatro medicamentos fueron a ensayos clínicos y fracasaron antes de que, en 1961, se decidieran por uno llamado ácido propiónico 2-(4-isobutilfenil), que más tarde se convertiría en ibuprofeno. No era el más activo, pero era el menos tóxico y demostró ser eficaz.
La patente del ibuprofeno fue concedida a Boots en 1962 y fue aprobado como un medicamento de prescripción siete años después. No será hasta 1964 cuando los estudios mostraron que el ibuprofeno era 16-32 veces más potente como antinflamatorio, 30 veces más potente como analgésico y 20 veces más potente como antipirético que la aspirina.
En 1967, Upjohn Co (Estados Unidos) desarrolló ibuprofeno tras ser aprobado en la FDA con la marca Motrin. El producto se comercializó por primera vez en 1969 en Reino Unido como tratamiento antirreumático con la marca Brufen.
Con los estudios adicionales de eficacia y seguridad, se convierte en producto OTC (Over The Counter o Especialidad Farmacéutica Publicitaria), siendo el primer AINE aprobado en esta categoría, tanto en Inglaterra (1983) como en Estados Unidos (1984), para el tratamiento del dolor y la fiebre.
Adams tardó 10 años en conseguir el éxito con el ibuprofeno “I did think we would succeed eventually- I always felt we would succeed”; de hecho, él mismo actúo de cobaya testando 2 o 3 compuestos.
Definición (39-41)
Los AINE forman una gran familia farmacológica heterogénea, pero todos tienen una cosa en común: la inhibición de una de las dos COX. Estas enzimas están involucradas en la cascada de ácido araquidónico, lo que conduce a una disminución en la síntesis de PG.
Los AINE generalmente se dividen en grupos según su estructura química y selectividad: salicilatos acetilados (aspirina), salicilatos no acetilados (diflunisal, salsalato), ácidos propiónicos (naproxeno, ibuprofeno), ácidos acéticos (diclofenaco, indometacina), ácidos enólicos (meloxicam, piroxicam), ácidos antranílicos (meclofenamato, ácido mefenámico), naftilalanina (nabumetona) e inhibidores selectivos de la COX-2 (celecoxib, etoricoxib) (42).
Clasificación (39-41)
Existen varias formas de clasificar a este grupo de fármacos. En general se usa la clasificación por estructura química (Figura 1) y la clasificación según su selectividad sobre las enzimas COX (Figura 2).
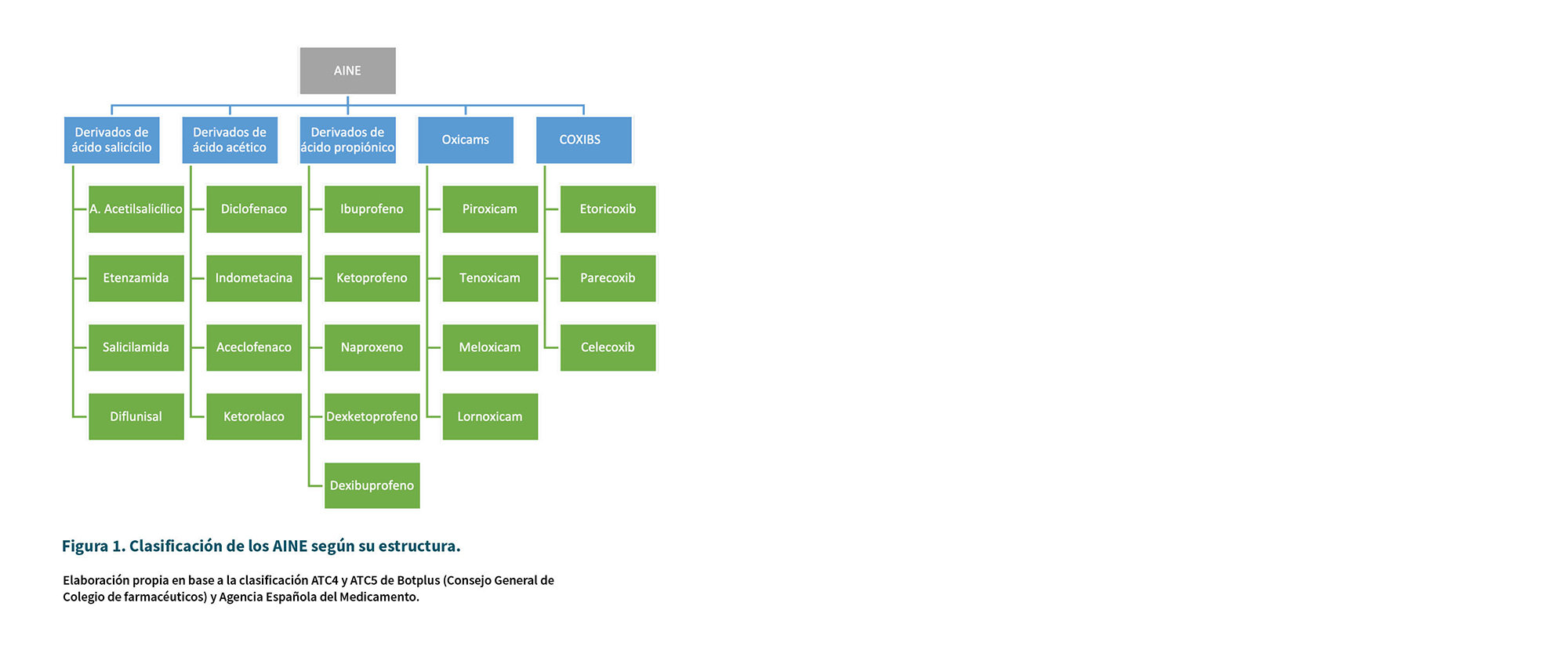
El paracetamol no es en sí mismo un fármaco antinflamatorio, ya que no tiene este efecto terapéutico, pero desde el punto de vista de mecanismo de acción podría incluirse también en esta clasificación, ya que se ha visto que actúa sobre la enzima COX-3.

Mecanismo de acción
El principal mecanismo de acción (39-41) de los AINE es la inhibición de la enzima COX. Esta enzima cataliza la síntesis de PG a partir del ácido araquidónico (AA) y de otros ácidos grasos precursores.
La COX es una enzima microsómica que aparece en forma de dímero (dos moléculas unidas para formar una unidad funcional) ubicada en la luz y la membrana del retículo endoplasmático. Las COX son parte de las enzimas bifuncionales, prostaglandin-endoperoxido-H sintetasa (PGHS, o prostaglandin endoperoxide-H synthase). La Figura 3 resume el mecanismo de acción de los AINE, así como los procesos de síntesis en los que participan las enzimas COX.
Los AINE reducen la actividad COX principalmente mediante la inhibición competitiva salvo el AAS, que ejerce una inhibición covalente e irreversible de COX. Las moléculas del ácido acetilsalicílico entran en el canal activo de la COX y añaden un grupo acetilo a un grupo R de un aminoácido del sitio activo de la COX. El grupo acetilo se une a la serina de la posición 530 de la COX-1 y a la serina de la posición 516 de la COX-2. Esto produce una inhibición irreversible o no competitiva.
El resultado final de esta inhibición COX es la reducción de la producción de PG y de otras moléculas.
Mecanismo antipirético
El desarrollo de la fiebre se debe a que las toxinas bacterianas y otros pirógenos estimulan la producción de citoquinas por parte de los leucocitos. Estas citoquinas aumentan la síntesis de PG en el área del hipotálamo modificando los niveles “termostato” del cuerpo. La vasodilatación cutánea asociada a las PG aumenta también la temperatura corporal.
Todos los AINE reducen la fiebre inhibiendo la síntesis de PG en el hipotálamo.
La Figura 3 resume la acción COX en el metabolismo del AA, así como los mecanismos de acción de los AINE (43). La conversión de AA implica la primera ciclación a 15-hidroxiperóxido inestable (PGG2) por la COX, y luego la actividad de la peroxidasa (POX) reduce el PGG2 generado a PGH2. La PGH sufre una mayor transformación en tejidos, generando diferentes reguladores endógenos: prostaglandinas de las series D (PGD2), E (PGE2) y F (PGF2), prostaciclina (PGI2) y tromboxano (TXA) (43). Estos prostanoides (lípidos bioactivos como PG y TX) interactúan con receptores específicos de membrana de la superfamilia proteína-G-acoplados, participando en numerosos procesos fisio-patológicos como la modulación de la inflamación y su resolución, la erosión del hueso, protección-ulceración gástrica, angiogenesis-cáncer, hemostasia-trombosis, hemodinámica renal-progresión de enfermedad renal, ateroprotección-progresión de aterosclerosis (44).
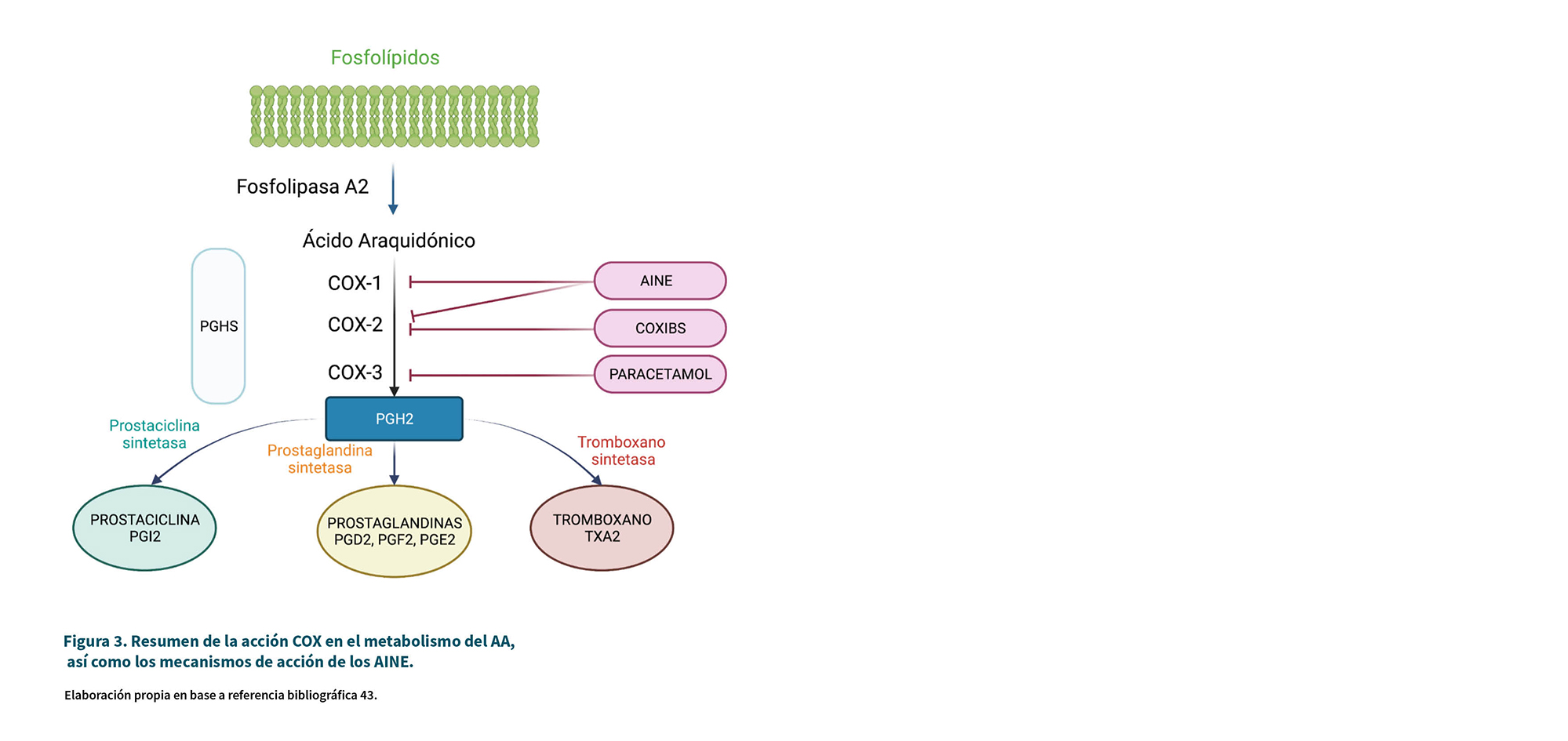
Hasta el momento se han identificado 3 isoenzimas: COX-1, COX-2, COX-3. Esta última fue descrita en 2002 y ha sido también recientemente asociada al mecanismo de acción del paracetamol (43):
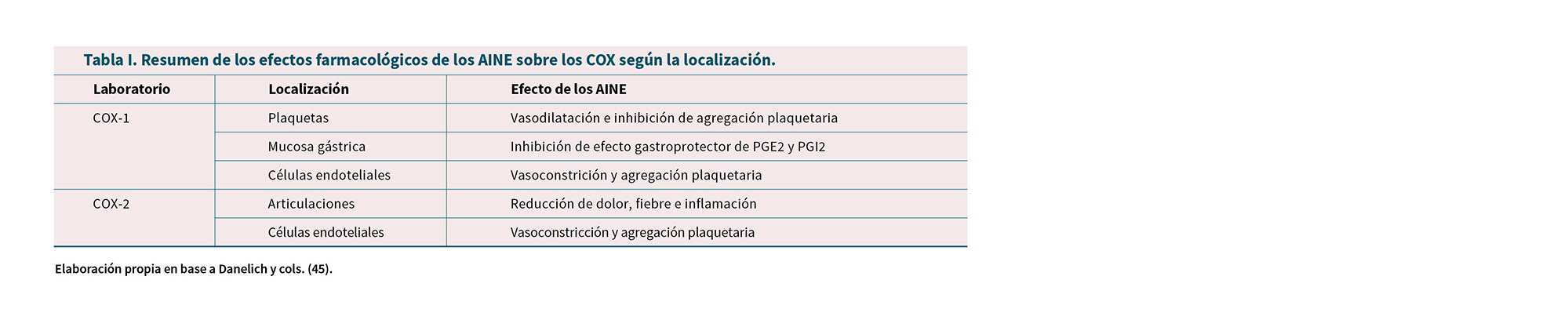
— COX-1: está continuamente expresado en los tejidos, principalmente en la mucosa gástrica y los riñones. La ingesta frecuente de AINE que actúan sobre esta enzima reduce la producción de PGE2 y PGI2 protectoras de la mucosa gástrica, lo que genera los efectos adversos de estos fármacos en forma de úlceras gástricas.
— COX-2: su actividad se registra solo durante los procesos de inflamación. Los COXIBS que actúan sobre esta enzima tienen menos efectos adversos a nivel gástrico que los que actúan sobre COX-1. Sin embargo, el COX-2 es la enzima de preferencia en los vasos sanguíneos para la síntesis de PGI2 que protege frente a la isquemia vascular. De forma que los fármacos selectivos COX-2 se asocian a efectos adversos cardiovasculares, como el aumento de la enfermedad cardiovascular y la isquemia.
— COX-3: aunque parece que se transcribe a partir del mismo gen que el COX-1, produce diferentes polipéptidos altamente sensibles a los fármacos analgésicos-antipiréticos, pero con bajo efecto antinflamatorio. Se cree que la retención del intrón 1 en su RNAm (respecto al COX-1) causa una inserción de 30 amino-ácidos en el péptido resultante, aunque estos aspectos están todavía en investigación. Parece que la COX-3 tiene actividad COX diferente de las otras 2, aunque más similar al COX-1. El COX-3 se expresa con más intensidad en la glándula pituitaria y el hipotálamo, lo que parece coherente con los efectos antipiréticos del paracetamol.
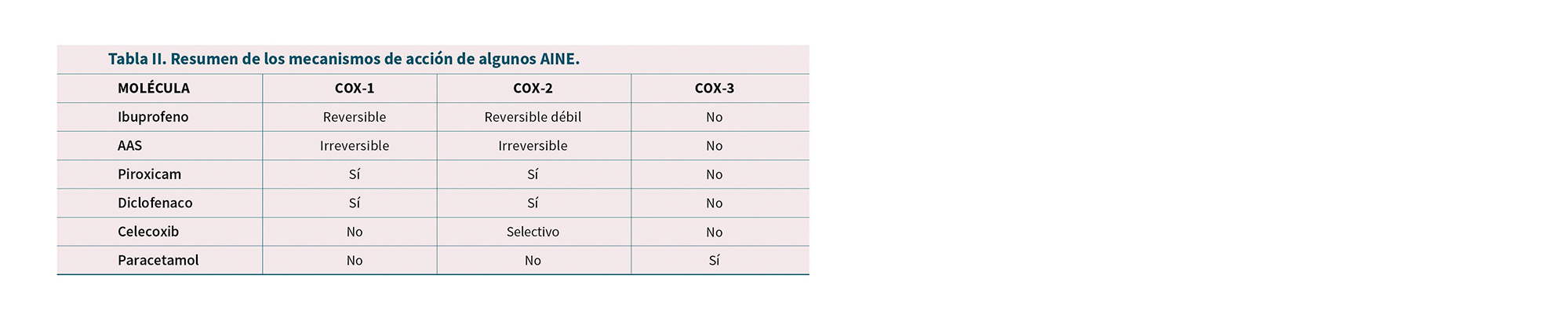
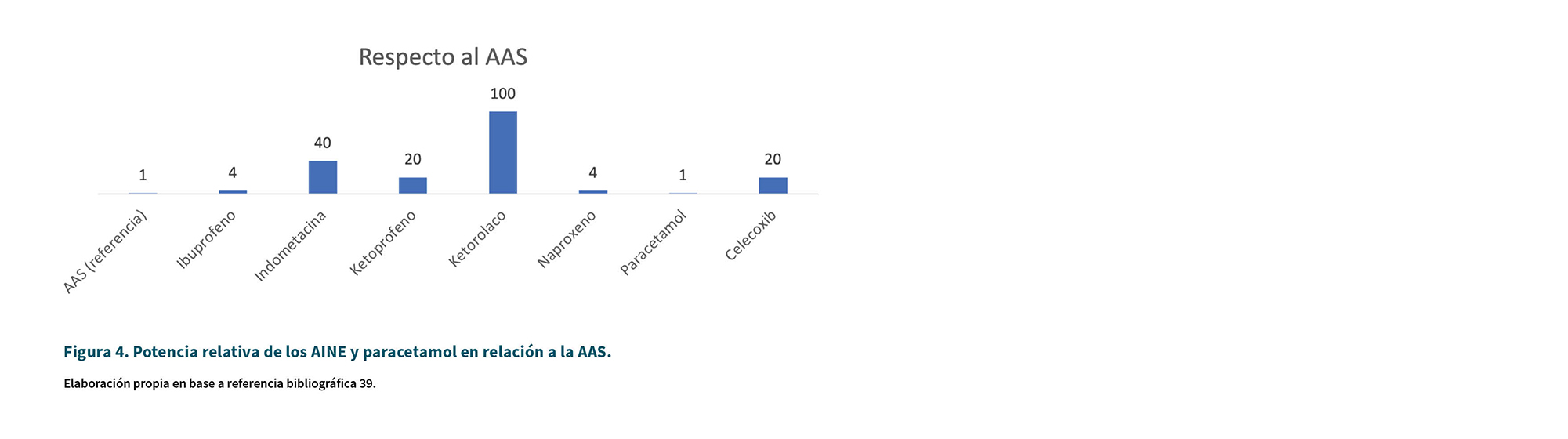
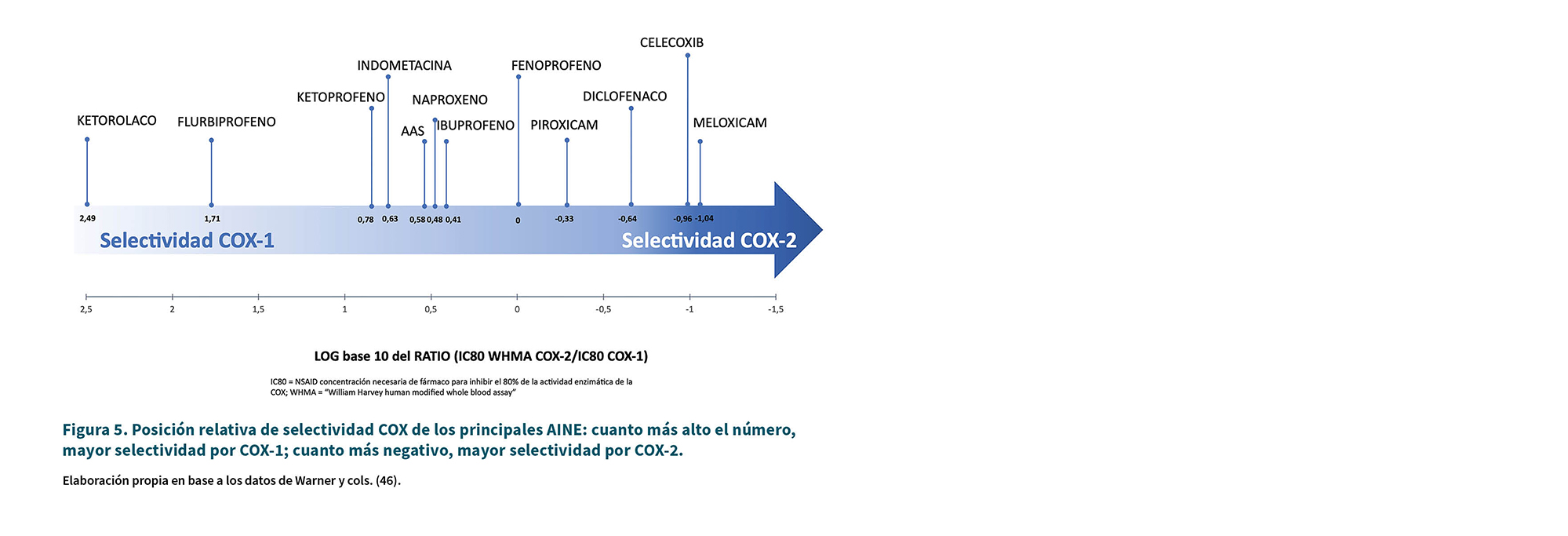
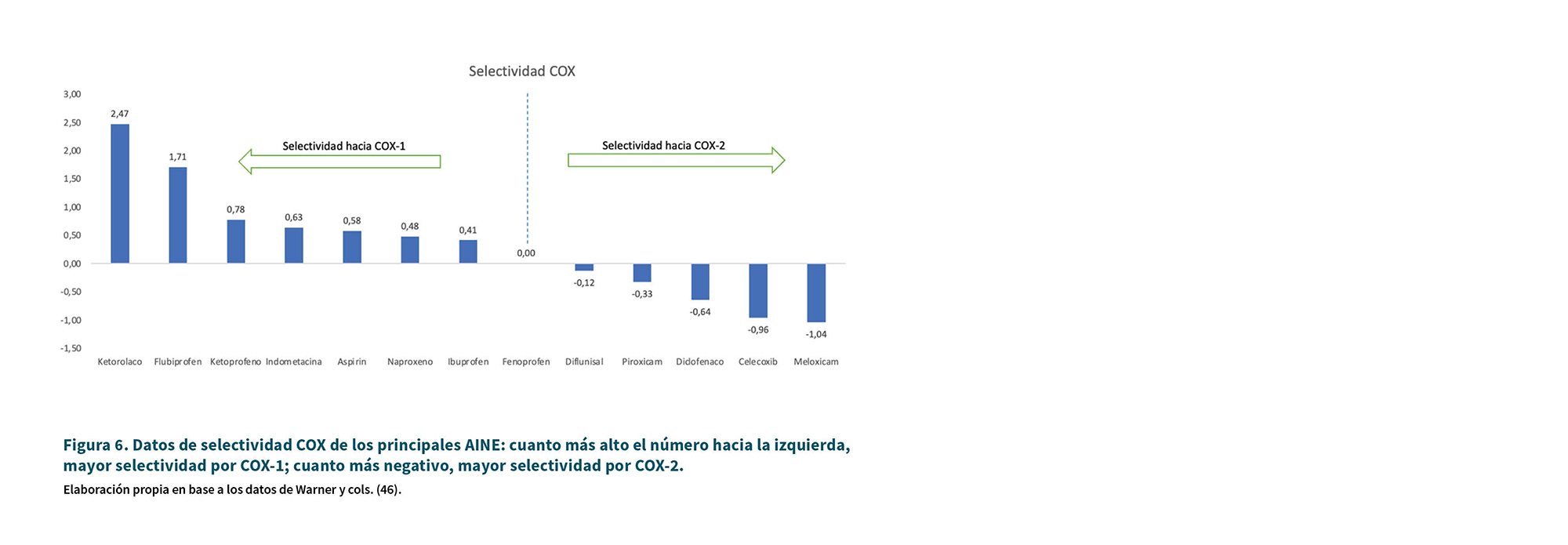
La Tabla I resume la ubicación de las enzimas COX y los efectos farmacológicos de los AINE que actúan sobre cada enzima COX (45). La Tabla II resume los mecanismos de acción de los AINE más importantes en relación a COX. La Figura 4 indica la potencia relativa de inhibición respecto a AAS mientras que las Figuras 5 y 6 muestran la selectividad COX de los principales AINE en base a los datos de Warner y cols. (46).
Efectos terapéuticos
Los efectos terapéuticos (39-41) de los AINE son múltiples y todavía están en estudio. Los más importantes y confirmados con evidencia científica se explican a continuación.
Efecto analgésico
Los AINE reducen el dolor causado por daño tisular o por los mediadores inflamatorios que actúan sobre las terminaciones nerviosas. Esta acción es indirecta, ya que actúan reduciendo las PG que sensibilizan las terminaciones nerviosas.
Efecto antipirético
La reducción de la fiebre es una característica de los AINE y del paracetamol. La temperatura corporal está controlada por el termostato hipotalámico. La fiebre se produce por la liberación de IL-1, PGE, entre otros mediadores, que alteran y elevan el valor de referencia de este termostato hipotalámico. Los AINE actúan inhibiendo esta liberación de PG.
Efecto antinflamatorio
El efecto antinflamatorio procede principalmente de su acción sobre las PG.
Efecto anticoagulante
Los AINE interfieren con el proceso de activación de las plaquetas al inhibir la acción de COX plaquetario bloqueando la activación de tromboxano A2 (47,48).
El AAS acetila irreversiblemente la COX-1 plaquetaria y tiene efecto más duradero sobre la síntesis de tromboxanos que otros salicilatos. El efecto antiagregante plaquetario de AAS persiste aproximadamente 14 días, mientras que en los demás AINE es mucho más corto. El tromboxano favorece la agregación plaquetaria mientras que la PGI2 la reduce.
El AAS puede inhibir de forma irreversible la producción de tromboxano en las plaquetas por acetilación de la COX-1. Este efecto es prolongado, debido a que las plaquetas carecen de núcleo y no sintetizan nueva COX, por lo que la falta de tromboxano dura toda la vida de la plaqueta (7-10 días) (47). Tras la interrupción del AAS, la actividad COX se recupera lentamente a medida que se producen nuevas plaquetas con COX intacto, es decir, que sus efectos sobre las plaquetas duran varios días.
La administración a largo plazo de AAS a dosis muy bajas (20 mg/día) es suficiente para bloquear la acción del tromboxano A2 y la inhibición de la agregación plaquetaria en más de un 95 % (47). Por ejemplo, tras una única dosis de aspirina los efectos antiplaquetarios se manifiestan a las 2 horas y persisten de 4 a 7 días.
El resto de AINE, que también inhiben el COX, ejercen una inhibición reversible y por tanto la inhibición de la acción plaquetaria es dependiente de la eliminación del fármaco en la circulación (47). En este caso, al ser la inhibición reversible, los efectos antiagregantes y en el tiempo de sangrado vendrán determinados por la vida media del fármaco. De este modo, los efectos antiagregantes de piroxicam persistirán varios días tras interrumpir el fármaco mientras que, con el ibuprofeno, el efecto desaparece a las 12 h. Dosis bajas de ibuprofeno (200 mg x 3 al día) no llegarían a prolongar el tiempo de sangrado mientras que dosis más altas pueden llegar a prolongarlos durante varias horas (47).
Por último, se ha de tener en cuenta que la administración concomitante de AAS con otro AINE puede impedir la acción antiplaquetaria del primero. Los AINE se unen de forma reversible a la COX plaquetaria de forma que reducirían la unión de AAS y, por tanto, la acción antiplaquetaria. La evidencia ha demostrado que la administración de ibuprofeno contrarresta la acción antiplaquetaria del AAS (49,50). La evidencia de esta interacción entre AAS y el resto de AINE es extensa (50,51).
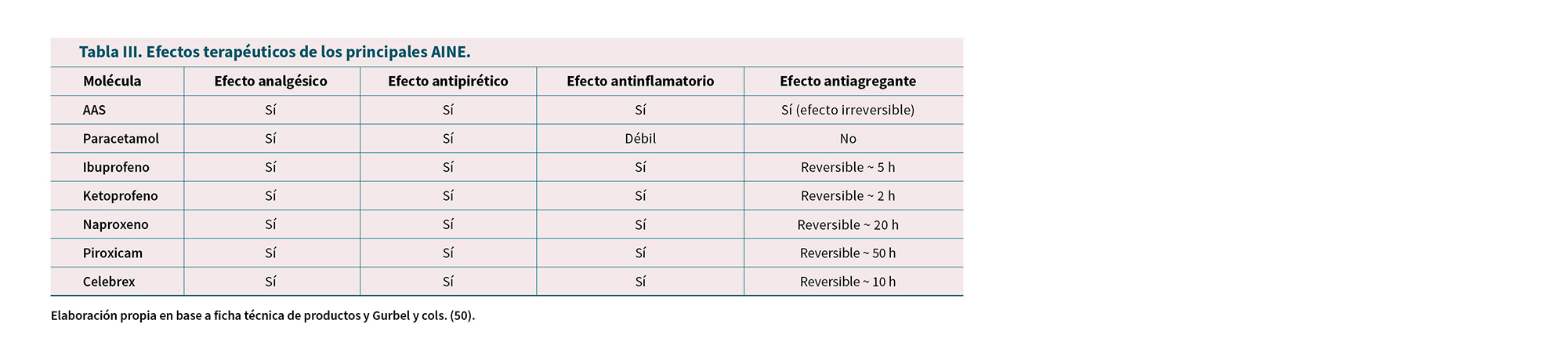
La Tabla III resume los efectos terapéuticos de los principales AINE.
Ácido acetilsalicílico
Antinflamatorio, analgésico, antipirético y antiagregante. Inhibe la COX reduciendo la formación de PG, lo que impide la estimulación de los receptores de dolor a nivel periférico (52); adicionalmente tiene efectos centrales a nivel del hipotálamo (52). La inhibición de la síntesis de PG explica los efectos analgésicos y antipiréticos.
Los efectos del AAS son dosis-dependiente (Figura 7).

Derivados de ácido propiónico: ibuprofeno, ketoprofeno y naproxeno
Son inhibidores no selectivos de la COX y ejercen acción analgésica, antipirética y antinflamatoria. Pueden alterar la función plaquetaria y alargar el tiempo de sangría. Son inhibidores reversibles.
El ibuprofeno inhibe de forma competitiva el efecto antiplaquetario del AAS si se administra de forma concomitante.
Derivados de ácido acético: indometacina
Tiene efecto antinflamatorio, analgésico y antipirético. Actúan por inhibición reversible de COX.
La indometacina es derivado de ácido indolacético, inhibidor potente de COX. Tiene más tendencia a causar efectos adversos, por lo que suele ser fármaco de reserva para situaciones agudas moderadas a graves. También se usa para tratar conducto arterial permeable en lactantes, ya que inhibe la síntesis de PG y lleva al cierre del conducto.
Su perfil de seguridad limita el uso crónico.
Oxicams: pirixocam, meloxicam
Antinflamatorio con potencia similar al naproxeno o AAS. El meloxicam inhibe COX-1 y COX-2 pero con más preferencia por COX-2. Pero a dosis altas es un AINE no selectivo.
Piroxiam presenta efecto analgésico, antipirético y antinflamatorio. Actúa también inhibiendo la agregación neutrofílica, la migración de polimorfonucleares y monocitos a la zona inflamada, así como la liberación de enzimas lisosómicas por parte de los leucocitos. Reduce la producción de factor reumatoide tanto sistémico como sinovial (53).
Ácidos heteroarilacéticos: diclofenaco, ketorolaco
El diclofenaco es más potente que la indometacina o el naproxeno. Tiene marcada actividad antinflamatoria, analgésica y antipirética (54). El ketorolaco tiene actividad analgésica potente. Se usa a corto plazo en dolor moderado como postoperatorio, hasta 5 días. Evitar su uso en pediatría y no superar dosis de 40 mg/día.
Inhibidores selectivos: celecoxib, etoricoxib, parecoxib
Efecto analgésico, antipirético y antinflamatorio, pero no inhiben la agregación plaquetaria porque las plaquetas solo tienen COX-1. Tienen inhibición selectiva COX-2. El celecoxib es 10-20 veces más potente sobre COX-2 que sobre COX-1 y a dosis terapéuticas no se observa inhibición estadísticamente significativa de COX-1 (55). No afecta el tromboxano plaquetario, por lo que carece de actividad anticoagulante a dosis recomendadas.
Es tan eficaz como el naproxeno en artrosis, pero parece insuficiente para controlar el dolor postoperatorio.
Tienen actividad antinflamatoria potente sin producir toxicidad digestiva significativa. El celecoxib fue el primero en comercializarse. En base a resultados de estudios en cáncer de colon, las dosis de 400 mg y 200 mg dos veces al día reportaron un aumento del riesgo de episodios cardiovasculares versus placebo de 3,4 y 2,5 veces, respectivamente. El producto sigue en el mercado, pero con advertencia de seguridad.
Hubo 2 COXIBS que fueron retirados del mercado al poco del lanzamiento por un aumento de los episodios cardiovasculares: rofecoxib fue retirado del mercado en 2004 por aumento del riesgo de infartoy accidente cerebrovascular versus placebo, y valdecoxib fue retirado del mercado en 2005 por efectos cardiovasculares y reacciones cutáneas graves.
Paracetamol
El paracetamol fue sintetizado por primera vez por Morse en 1878 y su primera utilización clínica la realizó Von Mering 15 años después. A finales los años 40 se confirmó que el paracetamol era el metabolito activo de la fenacetina, que tuvo que ser retirada por nefrotoxicidad. Posteriormente, a mediados de la década de los 50, se demostró su eficacia y seguridad. En España, la primera especialidad oral de paracetamol no apareció hasta el año 1974. En algunos países también se le denomina acetaminofen (56).
Está disponible desde hace 100 años y conocido como APAP (N-acetil-p-aminofenol).
Tiene efectos analgésicos y antinflamatorios a dosis tolerables, pero su efecto antinflamatorio es débil debido a que es inactivado por los peróxidos que se generan en el tejido inflamado.
Se desconoce exactamente el mecanismo de acción del paracetamol, aunque se sabe que actúa a nivel del SNC y, en menor grado, bloqueando la generación del impulso doloroso a nivel periférico (57).
Parece que el paracetamol tiene poco efecto sobre COX-1 y COX-2 (motivo por el que tiene poco efecto antinflamatorio) pero ejerce una inhibición de COX-3 (todavía en estudio) a nivel del SNC (que se asocia al dolor y la fiebre) pero no inhibe de forma significativa las COX en tejidos periféricos (57). La acción antitérmica se asocia a la inhibición del a PEG1 en el hipotálamo.
El paracetamol estimula la actividad de las vías serotoninérgicas descendentes que bloquean la transmisión de las señales nociceptivas a la médula espinal procedentes de tejidos periféricos (57).
Tampoco inhibe la síntesis de tromboxano ni la agregación plaquetaria, por lo que no tiene ninguna utilidad terapéutica en el campo cardiovascular.
No puede considerarse un miembro de los AINE, fundamentalmente por casi no tener el efecto antinflamatorio, aunque sí tiene efecto analgésico y antipirético. No afecta a la agregación plaquetaria ni prolonga el tiempo de coagulación de la sangre.
Sin embargo, algunos autores lo incluyen entre los AINE por su mecanismo de acción inhibidor de la COX-3.
Inhibe la síntesis de PG a nivel del SNC pero tiene efecto menor sobre las COX en tejidos periféricos, lo que explica el débil poder antinflamatorio.
Farmacocinética
La Tabla IV resume los parámetros farmacocinéticos de los AINE. Describimos a continuación los aspectos farmacocinéticos más relevantes de cada fármaco (39-41).
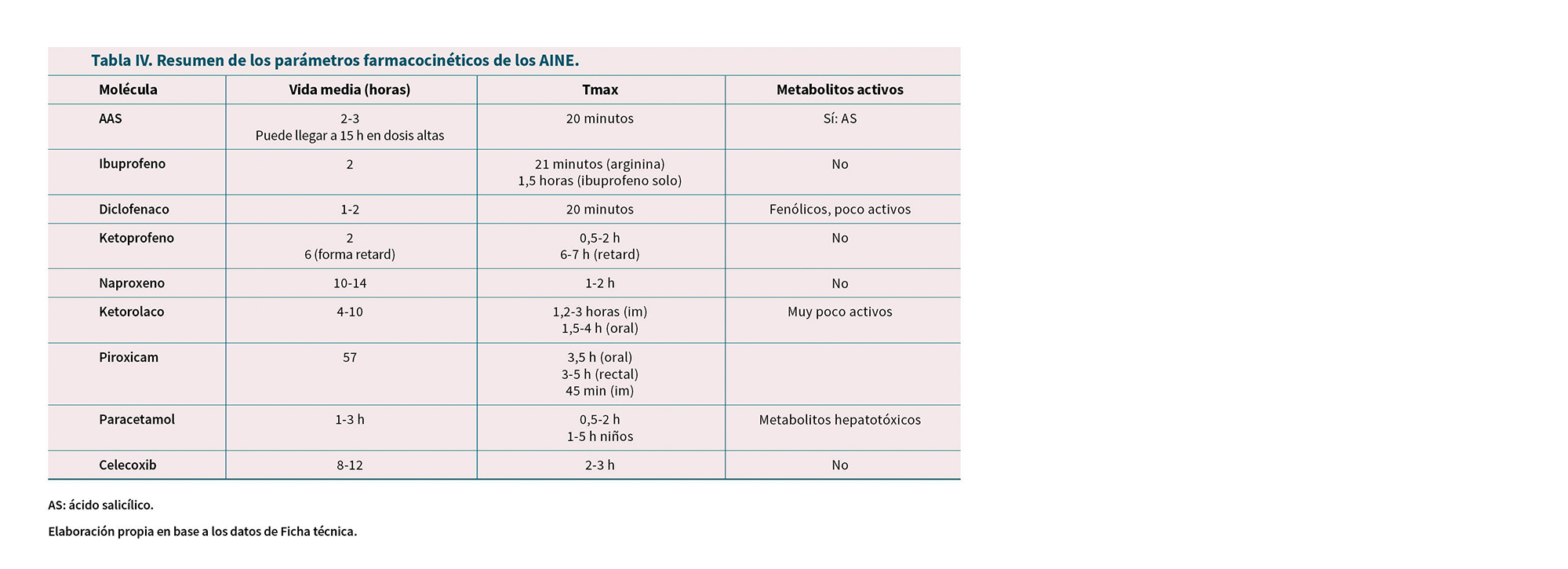
Ácido acetilsalicílico
Se absorbe bien en el intestino. La administración junto a antiácidos puede ralentizar la velocidad de absorción, pero no reduce de forma significativa la biodisponibilidad.
Es hidrolizado rápidamente a ácido salicílico (AS o salicilato) por la esterasa plasmática y esto explica su vida media
corta (15 minutos). Los niveles plasmáticos se alcanzan a los 18-30 minutos para el AAS y 0,72-2 horas para el AS, aunque puede variar según la formulación (52).
Se distribuye unido a proteínas plasmáticas (80-90 %) especialmente albúmina. Atraviesa la placenta.
La mayoría de los efectos del AAS se atribuyen al salicilato, que tiene una vida media de 2 horas, sin embargo, el propio AAS es el responsable directo de la inhibición irreversible de la COX plaquetaria y de la antiagregación plaquetaria.
La mayor parte del AS se conjuga con glicina para formar ácido salicilúrico que se excreta en orina. La velocidad de excreción se ve afectada por el pH, por lo que en caso de sobredosis se alcaliniza la orina mediante la administración de bicarbonato sódico para incrementar la ionización y la eliminación del AS.
A dosis terapéuticas, la velocidad del metabolismo y la excreción del salicilato son proporcionales a la concentración plasmática. A dosis excesivas las vías de eliminación se saturan rápidamente y se genera una eliminación de orden cero con una vida media de 15 o más horas. Las dosis más altas elevan rápidamente la concentración de salicilato plasmático hasta valores tóxicos, especialmente en ancianos, por lo que este grupo es especialmente sensible a la toxicidad por AAS.
Ibuprofeno, ketoprofeno y naproxeno
Se distribuyen ampliamente y sufren metabolismo extenso hasta metabolitos inactivos en el hígado, antes de su excreción renal.
El naproxeno tiene vida media más larga (14 h) que el ibuprofeno o ketoprofeno (2 h), por lo que el primero se administra solo 2 veces al día versus de 2-4 al día de los otros dos.
El ibuprofeno tiene farmacocinética lineal hasta dosis de al menos 800 mg. Se absorbe en tracto gastrointestinal en un 80 %. La distribución es con fuerte unión a proteínas plasmáticas en un 99 %. Se metaboliza en el hígado por hidroxilación y carboxilación, dando metabolitos que carecen de actividad farmacológica. La eliminación es renal y es total al cabo de 24 horas. El 90 % se elimina en forma de metabolitos inactivos (58).
Diclofenaco54
Absorción completa y rápida. La concentración media se alcanza a las 2 horas. La mitad sufre un primer paso hepático. La distribución muestra una unión a proteínas plasmáticas del 99,7 %, principalmente albúmina, pasa al líquido sinovial obteniéndose Cmax a las 2-4 horas. Se metaboliza en hidroxilación y metoxilación, dando lugar a diversos metabolitos; la mayoría se convierten en conjugados glucurónidos. Los metabolitos fenólicos son biológicamente activos, pero menos que el diclofenaco. Se elimina el 60 % en la orina y el resto a través de las heces.
Oxicams: pirixocam, meloxicam
Piroxicam (53) tiene absorción rápida y completa, que puede retrasarse junto con el alimento. Tiene concentraciones plasmáticas proporcionales a la dosis y la Cmax se alcanza a las 3-5 horas. Su vida media de 50 h permite su administración en una dosis única diaria; la mayoría de pacientes alcanza niveles plasmáticos estables a los 7-12 días. La distribución es unida a proteínas plasmáticas en un 99 %. Se metaboliza en el hígado por hidroxilación y otras reacciones que dan lugar a metabolitos sin actividad antinflamatoria. Se elimina en orina y heces, siendo el doble en la orina.
Celecoxib
Se absorbe rápidamente en el intestino, alcanzando concentraciones máximas a las 2-3 horas con una Tmax de 4 h. Se distribuye unido a proteínas en un 97 %. Es metabolizado por CYP2C9 y los metabolitos identificados no tiene actividad sobre COX. La concentración plasmática se aumenta en un 100 % en mujeres mayores de 65 años. Los pacientes con insuficiencia hepática leve tienen incremento de Cmax de un 53 %, un 41 % en insuficiencia moderada y un 146 % en insuficiencia severa. Se excreta en heces y orina. Tiene una vida media de 8-12 h. Es un fármaco con una gran variabilidad interindividual. Las concentraciones plasmáticas del estado estacionario se alcanzan a los 5 días (55).
Paracetamol
Se absorbe rápidamente en el intestino. Tiene biodisponibilidad del 75-85 %. Alcanza la Cmax entre 0,5-2 horas. Se distribuye de forma extensa en tejidos periféricos y SNC con solo un 10 % de unión a proteínas plasmáticas (57).
Sufre un primer paso hepático con cinética lineal, que desaparece a dosis superiores a 2 g al día. Se metaboliza en el hígado en un 90-95 %. Se elimina principalmente en conjugado con ácido glucorónico y, menos, con ácido sulfúrico y cisteína. La vida media es de 1,5-3 horas y aumenta en caso de sobredosis, insuficiencia hepática y pacientes mayores y niños (57).
Con relación a la hepatotoxicidad (Figura 6), el CYP450 transforma una pequeña cantidad en un intermediario quinónico que puede ser hepatotóxico. A dosis terapéuticas, este intermediario es inactivado rápidamente por conjugación con glutatión, pero a dosis tóxicas se agota este glutatión hepático produciendo una acumulación del intermediario tóxico que causa necrosis hepática. En pacientes con sobredosis de paracetamol en riesgo de hepatotoxicidad, se les puede administrar acetilcisteína (compuesto sulhidrilo) que se conjuga con el intermediario quinónico y lo transforman en inofensivo, de forma que se pueda prevenir la lesión hepática.
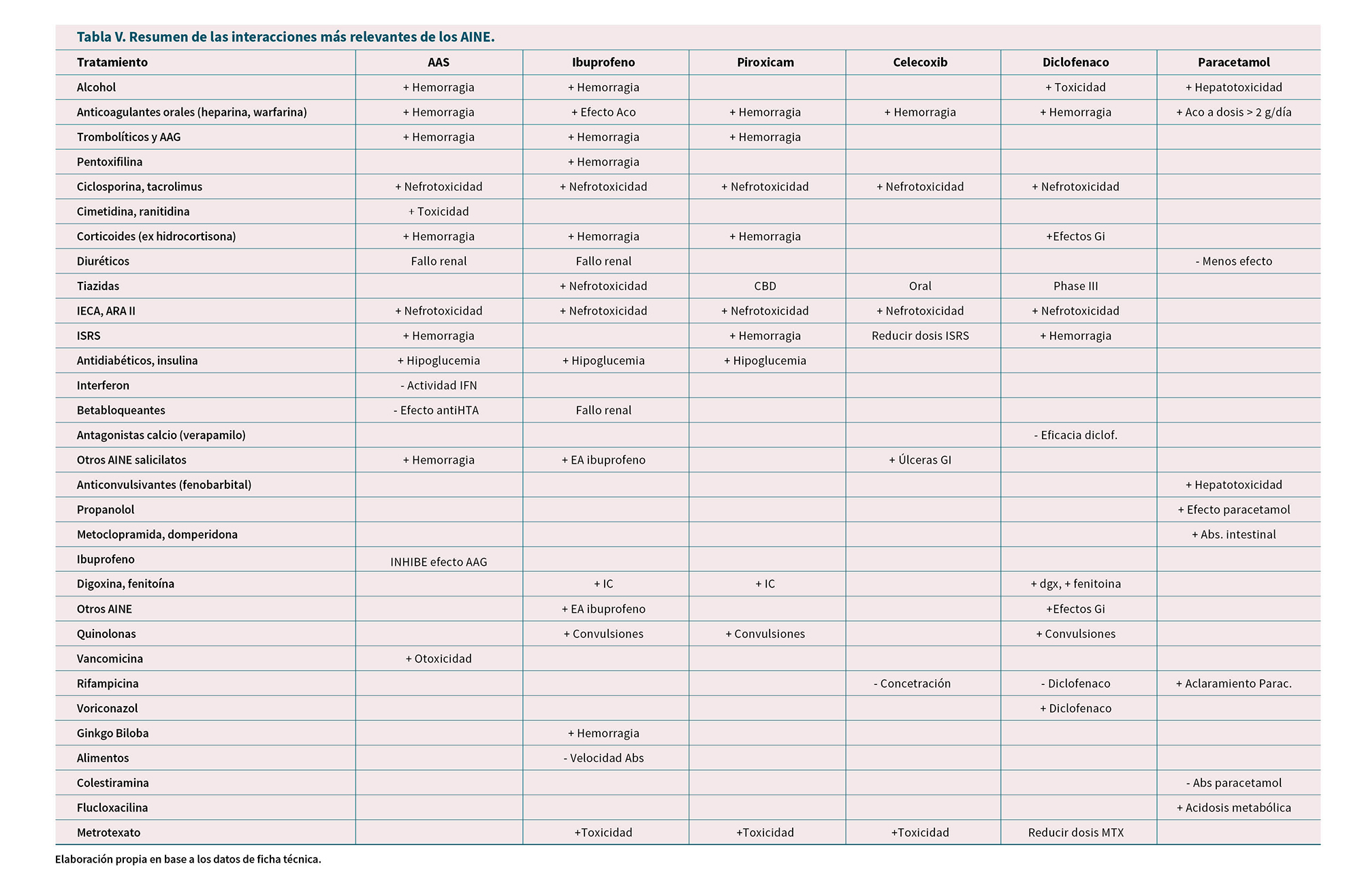
Interacciones
Los AINE puede interactuar con gran número de fármacos a través de la farmacocinética o farmacodinamia:
— La mayoría se excretan vía renal y pueden aumentar las concentraciones séricas y toxicidad del litio.
— Pueden reducir el aclaramiento del metotrexato y aminoglucósidos.
— Interferir en efecto antihipertensivo de diuréticos, agonistas beta-adrenérgicos, inhibidores de angiotensina y otros antihipertensivos.
— Cuando se administran junto a diuréticos ahorradores de potasio, pueden causar retención de potasio y consecuente hiperpotasemia.
— Dosis elevadas de salicilatos se asocian a efecto hipoglucémico que puede alterar los efectos de antidiabéticos.
— El ASA puede aumentar el efecto de la warfarina y provocar hemorragias.
— ASA interfiere en la acción de fármacos uricosúricos.
— La indometacina reduce el efecto natriurético de los diuréticos y puede causar nefrotoxicidad si se usa con triamtereno.
— Fluconazol, fluvastatina o similares pueden inhibir el metabolismo de celecoxib y aumentar la concentración sérica por lo que se deben reducir las dosis.
Pasamos a revisar las interacciones específicas de los 3 medicamentos más utilizados:
Ácido acetilsalicílico (52)
— Alcohol etílico: aumenta el riesgo de hemorragia digestiva.
— Anticoagulantes orales, como heparina y warfarina, aumenta el riesgo de hemorragia.
— Ibuprofeno: puede inhibir el efecto de dosis bajas de AAS sobre la agregación plaquetaria cuando se administran de forma concomitante, aunque no puede llegarse a conclusiones firmes y es probable que no haya un efecto clínicamente relevante con el uso ocasional de ibuprofeno.
— Ciclosporina: aumenta la nefrotoxicidad de la ciclosporina debido a efectos mediados por las prostaglandinas renales. Se recomienda una monitorización cuidadosa de la función renal, especialmente en pacientes ancianos.
— Cimetidina y ranitidina: la toxicidad del AAS se potencia con la administración de estas sustancias.
— Corticoides excepto hidrocortisona: puede incrementar el riesgo de úlceras y de hemorragias gastrointestinales, debido a un efecto sinérgico.
— Diuréticos: puede ocasionar un fallo renal agudo, especialmente en pacientes deshidratados.
— Inhibidores de la enzima convertidora de la angiotensina (ECA) y antagonistas de los receptores de la angiotensina II: efecto sinérgico en la reducción de la filtración glomerular, que puede ser exacerbado en caso de alteración de la función renal. La administración de esta combinación a pacientes ancianos o deshidratados puede llevar a un fallo renal agudo. Se recomienda una monitorización de la función renal. También puede reducir el efecto antihipertensivo de los inhibidores de la ECA.
— Inhibidores selectivos de la recaptación de serotonina: aumenta el riesgo de hemorragia en general y digestiva alta en particular.
— Antidiabéticos como insulina y sulfonilureas: aumenta el efecto hipoglucemiante por desplazamiento de los receptores de las proteínas plasmáticas.
— Interferón: disminuye la actividad.
— Otros antihipertensivos (beta-bloqueantes): disminuir el efecto antihipertensivo debido a una inhibición de las PG con efecto vasodilatador.
— Otros AINE con salicilatos: puede incrementar el riesgo de úlceras y de hemorragias gastrointestinales, debido a un efecto sinérgico.
— Trombolíticos y antiagregantes plaquetarios: aumenta el riesgo de hemorragia, por lo que no se recomienda.
— Vancomicina: aumenta el riesgo de otoxicidad.
Ibuprofeno (58)
— Ácido acetilsalicílico: no se recomienda la administración concomitante de debido a la posibilidad de que aumenten los efectos adversos, ya que el ibuprofeno puede inhibir de forma competitiva el efecto de dosis bajas de AAS sobre la agregación plaquetaria cuando se administran de forma concomitante.
— Antihipertensivos (incluidos los inhibidores de la ECA, antagonistas de la angiotensina II y los betabloqueantes) y los diuréticos: inhiben la COX y puede resultar en un mayor deterioro de la función renal, posible insuficiencia renal aguda, que normalmente es reversible. Adecuar la hidratación y monitorización.
— Anticoagulantes: los AINE pueden aumentar los efectos de los anticoagulantes tipo dicumarínico, como la warfarina; deberá evitarse el uso simultáneo.
— Antiagregantes plaquetarios: aumentan el riesgo de hemorragia gastrointestinal y no deben combinarse con ticlopidina, debido al riesgo de un efecto aditivo en la inhibición de la función plaquetaria.
— Corticoides: aumenta el riesgo de úlcera o sangrado gastrointestinal.
— Inhibidores selectivos de la recaptación de serotonina (ISRS): aumenta el riesgo de sangrado gastrointestinal.
— Otros AINE: aumenta el riesgo de aparición de reacciones adversas gastrointestinales (úlcera gastrointestinal y hemorragias).
— Metotrexato administrado a dosis de 15 mg/semana o superiores dentro de un intervalo de 24 horas: aumento del nivel plasmático de metotrexato por reducción del aclaramiento renal. Debe evitarse el ibuprofeno en pacientes que reciban tratamiento con metotrexato a dosis elevadas. En el metotrexato administrado a dosis bajas, inferiores a 15 mg/semana, se vigilarán estrechamente los valores hemáticos del paciente.
— Sulfamidas: puede aumentar sus efectos tóxicos.
— Digoxina, fenitoína: puede aumentar sus niveles séricos.
— Digoxina: puede exacerbarse la insuficiencia cardiaca; reducir la tasa de filtrado glomerular.
— Pentoxifilina: puede aumentar riesgo de hemorragia.
— Quinolonas: puede aumentar riesgo convulsiones.
— Tiazidas: se contrarresta el efecto diurético y el diurético puede aumentar la nefrotoxicidad del ibuprofeno.
— Hipoglucemiantes: aumenta el efecto hipoglucemiante de antidiabéticos orales e insulina. Es necesario ajustar dosis.
— Ciclosporina: posible riesgo toxicidad renal.
— Aminoglucósidos: se reduce su excreción.
— Ginkgo biloba: potencia el riesgo de hemorragia.
— Alcohol: aumenta riesgo de efectos adversos gastrointestinales incluyendo hemorragias.
— Alimentos: reducen la velocidad de absorción.
Paracetamol (57)
— Anticoagulantes orales: en dosis > 2 g/día de paracetamol se puede provocar un aumento de actividad anticoagulante, por reducción de síntesis hepática de factores de coagulación.
— Alcohol: potencia toxicidad hepática del paracetamol.
— Anticonvulsivantes (fenitoína, fenobarbital, metilfenobarbital, primidona): reducen la biodisponibilidad del paracetamol, potencian la hepatotoxicidad.
— Diuréticos de asa: tendrán menos efecto.
— Lamotrigina: se reduce su área bajo la curva y su vida media.
— Metoclopramida y domperidona: aumenta absorción intestinal de paracetamol.
— Propanolol: potencia la acción de paracetamol.
— Rifampicina: aumenta aclaramiento de paracetamol.
— Resinas como colestiramina: reducen absorción de paracetamol.
— Flucloxacilina: se puede producir acidosis metabólica.
El resumen de las principales interacciones a tener en cuenta se presenta en la Tabla V.
Efectos adversos
En este apartado repasaremos los efectos adversos (39-41) que se han de tener más presentes en cada fármaco. Hay que recordar que los AINE no están recomendados a partir de la segunda mitad del embarazo porque pueden tener efectos adversos sobre el feto debido a la inhibición de las PG (hemorragia digestiva, inhibición plaquetaria, disfunción renal y cierre prematuro del conducto arterial). Asimismo, el AAS debe evitarse, ya que la inhibición de la síntesis de PG puede ocasionar efectos adversos durante el embarazo y el desarrollo embriofetal. El paracetamol a dosis bajas puede usarse durante el embarazo bajo condiciones normales de uso y después de evaluación beneficio-riesgo (57).
El AAS debe evitarse durante el embarazo, ya que la inhibición de la síntesis de PG puede ocasionar efectos adversos durante el embarazo y el desarrollo embriofetal.
Ácido acetilsalicílico
— Síndrome de Reye: se debe evitar el uso de AAS en niños porque el riesgo de síndrome de Reye está incrementado en este grupo cuando están infectados por virus.
— Síntomas gastrointestinales: incluyendo irritación gástrica, hemorragia digestiva o úlceras pépticas. Se produce a dosis terapéuticas moderadamente elevadas que pueden producir acúfenos (sensación auditiva anormal, zumbido), que se considera un signo temprano de toxicidad por salicilatos.
— Hiperventilación: a dosis excesivas. Se debe a una estimulación directa e indirecta del centro respiratorio bulbar que lleva al aumento de la eliminación de CO2 y alcalosis respiratoria.
— Fiebre, deshidratación y acidosis metabólica grave: a dosis excesivas. Si no se trata rápidamente estos episodios puede llevar a Shock, coma, insuficiencia multiorgánica y muerte.
— Hipoprotombinemia: a dosis excesivas. Es una alteración de la hemostasia que causa hemorragia.
— Anafilaxia: es infrecuente, pero puede ser grave y potencialmente mortal. Se produce rinitis vasomotora, angioedema y urticaria. Se da principalmente en pacientes con asma, pólipos nasales o urticaria crónica. El paciente que ha tenido una reacción de hipersensibilidad a AAS no debe recibir ningún AINE, ya que existe una sensibilidad cruzada.
La sobredosis (el salicilismo) se puede producir con dosis de más de 100 mg/kg al día durante más de dos días (52). La toxicidad crónica puede ser de difícil diagnóstico por presentar signos y síntomas no específicos: mareos, vértigo, zumbidos, náuseas, vómitos, sordera, sudores, dolores de cabeza, confusión, vasodilatación, visión borrosa, entre otros (52). El zumbido de oídos se produce a concentraciones plasmáticas de 150 a 300 mcg/ml.
La intoxicación aguda se produce por la alteración del equilibrio ácido-base, se produce a concentraciones plasmáticas de 300 mg/l y los síntomas más frecuentes son náuseas, vómitos, acidosis metabólica, hiperventilación, asfixia, disritmias, hipotensión, los trastornos neurológicos como la confusión, delirio, convulsiones y coma, entre otros (52). El tratamiento de la sobredosis o intoxicación aguda por AAS debe incluir lo siguiente:
a) Inducción del vómito y lavado gástrico para extraer el fármaco no absorbido.
b) Administración i.v. de bicarbonato sódico para contrarrestar la acidosis metabólica, incrementar la ionización del salicilato en riñones y aumentar la velocidad de excreción del salicilato.
c) Administrar líquidos, electrolitos y otros tratamientos de soporte necesarios.
Ibuprofeno, naproxeno
— Producen irritación gástrica dependiente de la dosis. La administración prolongada de dosis altas se asocia a úlcera péptica, que se asocia también a náuseas, dispepsia, hemorragia.
— A dosis terapéuticas tiene pocos efectos adversos.
— Toxicidad hepática y renal. Se ha descrito insuficiencia renal aguda a dosis terapéuticas en pacientes deshidratados.
— Trastornos del sistema nervioso: fatiga, somnolencia, cefalea, mareo, vértigo.
— Erupción cutánea.
La mayoría de los casos de sobredosis han sido asintomáticos. Existe sintomatología a dosis superiores a 80-100 mg/kg, a las 4 h de la ingesta y generalmente son: dolor abdominal, náuseas, vómitos, diarrea, aturdimiento, cefalea y espasmos, entre otros. En caso de intoxicación grave se puede dar acidosis metabólica. En caso de cantidades importantes se debe realizar: lavado de estómago antes de 1 h.
Indometacina
— Efectos adversos digestivos y en el SNC superior al resto de AINE.
— Efectos adversos frecuentes: depresión, cefaleas, mareos, aturdimiento, vértigo, anorexia, náuseas, vómitos, dolor abdominal (59).
— Toxicidad hematológica grave: limitar uso a corto plazo y monitorizar a los pacientes.
— Edemas especialmente en pacientes con hipertensión o insuficiencia cardiaca.
Ketorolaco
— Toxicidad hematológica: uso limitado a 5 días o menos.
— Precaución en pacientes con insuficiencia renal o hepática: aumenta el riesgo de insuficiencia grave.
Celecoxib (55)
— Diarrea, dispepsia, dolor abdominal, náuseas y vómitos.
— Úlceras gastroduodenales, pero menor que los AINE no selectivos, como ibuprofeno o naproxeno.
— Hipertensión.
— Erupción y prurito.
— Efectos cardiovasculares: aumento de eventos graves como infarto de miocardio en pacientes con poliposis adenomatosa. Los pacientes con factores de riesgo cardiovascular deberán tratarse con precaución tras valoración.
— Toxicidad renal similar a otros AINE.
Paracetamol (57)
— El uso prolongado de paracetamol se asocia a un aumento de riesgo de disfunción renal.
— A dosis terapéuticas no presenta efectos tóxicos.
— En dosis tóxicas se produce inicialmente náuseas y vómitos y 24 h después la hepatotoxicidad. La sobredosis con paracetamol se evalúa en 4 fases:
• Fase I (12-24 h): náuseas, vómitos, diaforesis, anorexia.
• Fase II (24-48 h): mejoría clínica, se elevan AST, ALT, biblirrubina, protrobina.
• Fase III (72-96 h): pico de hepatotoxicidad.
• Fase IV (7-8 días): recuperación.
— La mínima dosis tóxica es 6 g en adultos y más de
100 mg/kg de peso en niños. Dosis superiores a 20-25 g son potencialmente fatales.
— La ingesta crónica de dosis superiores a 4 g/día da lugar a hepatotoxicidad transitoria.
El tratamiento de la sobredosis de paracetamol es la n-acetilcisteína (que actúa de antídoto), que puede prevenir o reducir significativamente la hepatotoxicidad, pero hay que tener en cuenta que esta avanza rápidamente a lo largo de los días tras la intoxicación. La efectividad del antídoto es máxima si se administra antes de que transcurran 4 h tras la intoxicación, y disminuye a partir de la octava hora, siendo ineficaz a partir de las 15 h.
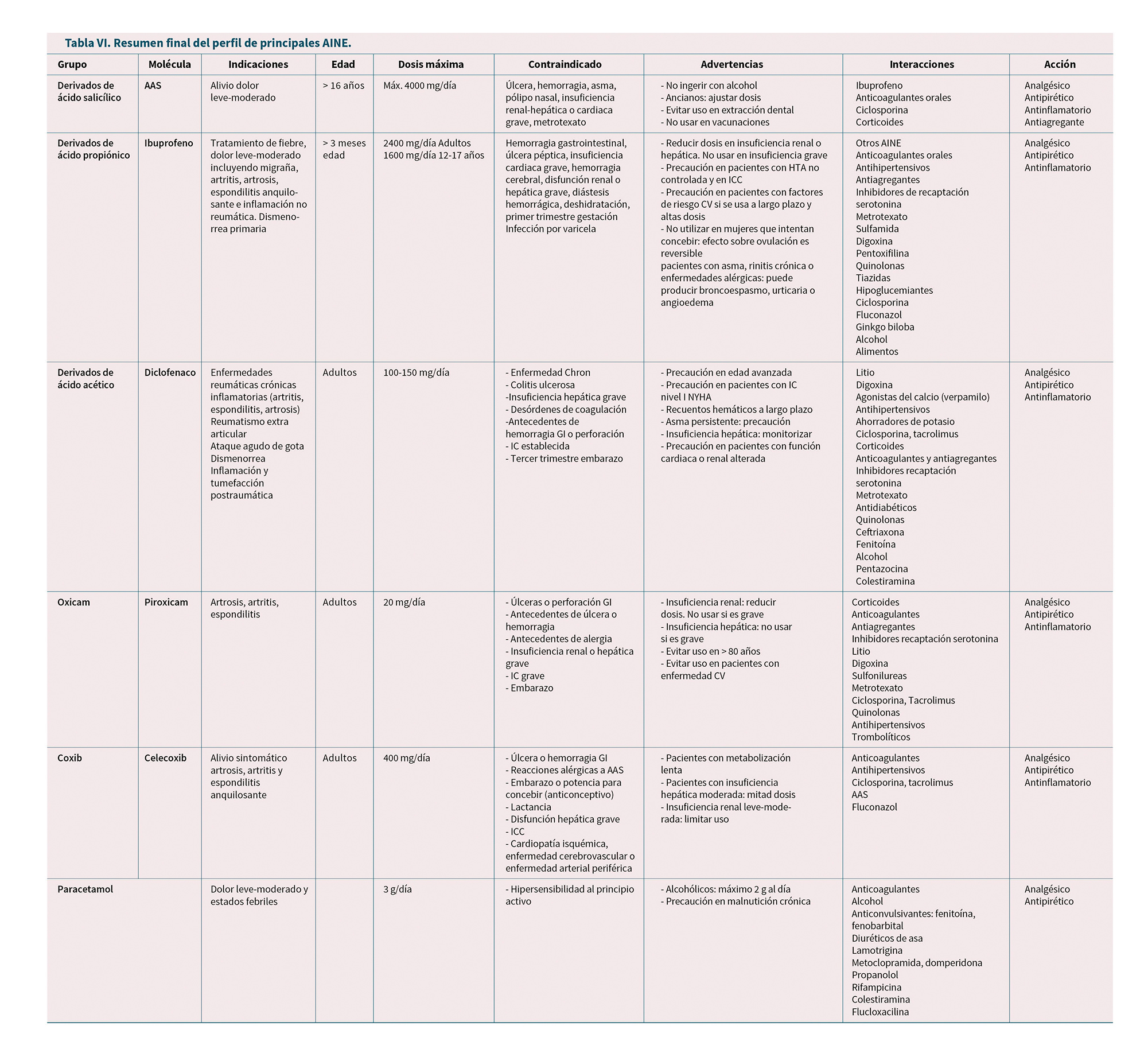
Para terminar, ofrecemos en la Tabla VI un resumen de las principales características de los AINE más utilizados.
Conclusiones
Los AINE con un grupo de fármacos heterogéneo, que tienen un mecanismo de acción común (inhibición de enzimas COX) pero que presentan distintos perfiles de selectividad COX, así como diferencias relevantes a nivel de espectro de acción terapéutica, perfil farmacocinético, tipos de interacciones y efectos adversos. Son fármacos altamente prescritos y es conveniente tener presentes estas diferencias a la hora de seleccionar el mejor fármaco según la indicación, características, polimediación y comorbilidades del paciente.
bibliografía
1. Mann J. Murder, Magic and Medicine. Oxford: Oxford University Press; 1992.
2. Bryan CP. The Papyrus Ebers. New York: Appleton; 1931.
3. Montinari MR, Minelli S, De Caterina R. The first 3500 years of aspirin history from its roots - A concise summary. Vascul Pharmacol. 2019;113:1-8. DOI: 10.1016/j.vph.2018.10.008.
4. Wood JN. From plant extract to molecular panacea: a commentary on Stone (1763) ‘An account of the success of the bark of the willow in the cure of the agues’. Philos Trans R Soc Lond B Biol Sci. 2015;370(1666):20140317. DOI: 10.1098/rstb.2014.0317.
5. Stone E. An account of the success of the bark of the willow in the cure of agues, Phil Trans. 1763;53:195-200.
6. Rigatelli B. Sostituto indigeno del solfato di chinina, Biblioteca Italiana o sia Giornale di Letteratura, Scienze ed Arti. 1824;33:267-71.
7. Vane JR, Botting RM. Aspirin and Other Salicylates. London, New York: Chapman & Hall Medical; 1992.
8. Leroux H. Découvert de la salicine. J de Chimie Medicale. 1830;6:341.
9. Piria R. Sur la composition de la salicine et quelques-unes de ses réactions. Comptes Rendues de l’Academie des Sciences, Paris. 1838;6:338.
10. Sneader W. Drug discovery - a history. Chichester: John Wiley & Sons; 2005.
11. Zundorf U. Aspirin 100 Years: The Future Has Just Begun, Leverkusen, Bayer AG, Consumer Care Business Group. Archives; 1997.
12. Sneader W. The discovery of aspirin: A reappraisal. BMJ. 2000;321:1591-4.
13. González Iglesias J. Historia de la Anestesia. Madrid: Editores Médicos SA; 1995.
14. Jeffreys D. Aspirin: The Remarkable Story of a Wonder Drug. 1.st ed. New York: Bloomsbury; 2004.
15. Witthauer K. Aspirin, ein neues Salicylpräparat. Die Heilkunde. 1899;3:396-8.
16. Wohlgemut J. Über Aspirin (Acetylsalicylsäure). Therap Mschr. 1899:276-8.
17. Floeckinger F. An experimental study of aspirin, a new salicylic acid preparation, Med News. 1899;75:641-72.
18. Flower RJ. The development of COX2 inhibitors. Nat Rev Drug Discov. 2003;2(3):179-91. DOI: 10.1038/nrd1034.
19. Sinniah A, Yazid S, Flower RJ. From NSAIDs to Glucocorticoids and Beyond. Cells. 2021;10(12):3524. DOI: 10.3390/cells10123524.
20. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231(25):232-5. DOI: 10.1038/newbio231232a0.
21. Botting RM. Vane’s discovery of the mechanism of action of aspirin changed our understanding of its clinical pharmacology. Pharmacol Rep. 2010;62(3):518-25. DOI: 10.1016/s1734-1140(10)70308-x.
22. Vane JR. Adventures and excursions in bioassay: the stepping stones to prostacylin. Biosci Rep. 2004;24(4-5):254-79. DOI: 10.1007/s10540-005-2734-7.
23. Hawkey CJ. COX-2 chronology. Gut. 2005;54(11):1509-14. DOI: 10.1136/gut.2005.065003.
24. Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A. 1993;90(24):11693-7. DOI: 10.1073/pnas.90.24.11693.
25. Cairns JA. The coxibs and traditional nonsteroidal anti-inflammatory drugs: a current perspective on cardiovascular risks. Can J Cardiol. 2007;23(2):125-31. DOI: 10.1016/s0828-282x(07)70732-8.
26. Chen J, Stark LA. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines. 2017;5(3):43. DOI: 10.3390/biomedicines5030043.
27. Dachineni R, Ai G, Kumar DR, Sadhu SS, Tummala H, Bhat GJ. Cyclin A2 and CDK2 as Novel Targets of Aspirin and Salicylic Acid: A Potential Role in Cancer Prevention. Mol Cancer Res. 2016;14(3):241-52. DOI: 10.1158/1541-7786.MCR-15-0360.
28. Ishihara T, Yoshida M, Arita M. Omega-3 fatty acid-derived mediators that control inflammation and tissue homeostasis. Int Immunol. 2019;31(9):559-67. DOI: 10.1093/intimm/dxz001.
29. Weng J, Zhao G, Weng L, Guan J; Alzheimer’s Disease Neuroimaging Initiative. Aspirin using was associated with slower cognitive decline in patients with Alzheimer’s disease. PLoS One. 2021;16(6):e0252969. DOI: 10.1371/journal.pone.0252969.
30. Jorda A, Aldasoro M, Aldasoro C, Guerra-Ojeda S, Iradi A, Vila JM, et al. Action of low doses of Aspirin in Inflammation and Oxidative Stress induced by aβ1-42 on Astrocytes in primary culture. Int J Med Sci. 2020;17(6):834-43. DOI: 10.7150/ijms.40959.
31. Flower R. What are all the things that aspirin does? BMJ. 2003;327(7415):572-3. DOI: 10.1136/bmj.327.7415.572.
32. Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(3A):2S-8S; discussion 21S-22S. DOI: 10.1016/s0002-9343(97)00203-9.
33. Brune K, Renner B, Tiegs G. Acetaminophen/paracetamol: A history of errors, failures and false decisions. Eur J Pain. 2015;19(7):953-65. DOI: 10.1002/ejp.621.
34. Graham GG, Scott K. Mechanism of action of paracetamol. Am J Ther. 2005;12(1):46-55. DOI: 10.1097/00045391-200501000-00008.
35. Prescott LF. Paracetamol: past, present, and future. Am J Ther. 2000;7(2):143-7.
36. Flower RJ, Vane JR. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature. 1972;240(5381):410-1. DOI: 10.1038/240410a0.
37. Dinchuk JE, Liu RQ, Trzaskos JM. COX-3: in the wrong frame in mind. Immunol Lett. 2003;86(1):121. DOI: 10.1016/s0165-2478(02)00268-7.
38. Dr. Stewart Sanders Adams (16 April 1923 to 30 January 2019) : Pioneer in the discovery of ibuprofen, that from meagre beginnings to become world’s best-selling pain-killing drug. Inflammopharmacology. 2019;27(1):1-4. DOI: 10.1007/s10787-019-00576-7.
39. Rang y Dale. Flashcards Farmacología. 2.ª ed. Madrid: Elservier; 2021.
40. Brenner G, Stevens C. Farmacología básica. 6.ª ed. Madrid: Elservier; 2019.
41. Page C. Farmacología esencial. 3.ª ed. Madrid: Elservier; 2022.
42. Ghlichloo I, Gerriets V. Nonsteroidal Anti-inflammatory Drugs (NSAIDs) [Updated 2022 May 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK547742/
43. Przybyła GW., Szychowski KA., Gmiński J. Paracetamol - An old drug with new mechanisms of action. Clin Exp Pharmacol Physiol. 2021;48(1):3-19. DOI: 10.1111/1440-1681.13392.
44. Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82(1-4):85-94. DOI: 10.1016/j.prostaglandins.2006.05.019.
45. Danelich IM, Wright SS, Lose JM, Tefft BJ, Cicci JD, Reed BN. Safety of nonsteroidal antiinflammatory drugs in patients with cardiovascular disease. Pharmacotherapy. 2015;35(5):520-35. DOI: 10.1002/phar.1584.
46. Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis [published correction appears in Proc Natl Acad Sci U S A. 1999;96(17):9666]. Proc Natl Acad Sci U S A. 1999;96(13):7563-8. DOI: 10.1073/pnas.96.13.7563.
47. Driver B, Marks DC, van der Wal DE. Not all (N)SAID and done: Effects of nonsteroidal anti-inflammatory drugs and paracetamol intake on platelets. Res Pract Thromb Haemost. 2019;4(1):36-45. DOI: 10.1002/rth2.12283.
48. Schafer AI. Effects of nonsteroidal anti-inflammatory therapy on platelets. Am J Med. 1999;106(5B):25S-36S. DOI: 10.1016/s0002-9343(99)00114-x.
49. Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345(25):1809-17. DOI: 10.1056/NEJMoa003199.
50. Gurbel P, Tantry U, Weisman S. A narrative review of the cardiovascular risks associated with concomitant aspirin and NSAID use. J Thromb Thrombolysis. 2019;47(1):16-30. DOI: 10.1007/s11239-018-1764-5.
51. Saxena A, Balaramnavar VM, Hohlfeld T, Saxena AK. Drug/drug interaction of common NSAIDs with antiplatelet effect of aspirin in human platelets. Eur J Pharmacol. 2013;721(1-3):215-24. DOI: 10.1016/j.ejphar.2013.09.032.
52. Ficha técnica de Aspirina 500 mg [Internet]. Agencia Española del Medicamento; julio de 2022. Disponible en: https://cima.aemps.es/cima/dochtml/ft/67268/FT_67268.html
53. Ficha técnica de Feldene 20 mg (piroxicam) [Internet]. Agencia Española del Medicamento; marzo de 2023. Disponible en: https://cima.aemps.es/cima/pdfs/es/p/55806/55806_p.pdf
54. Ficha técnica de Diclofenaco cinfa 50 mg [Internet]. Agencia Española del Medicamento; marzo de 2023. Disponible en: https://cima.aemps.es/cima/dochtml/p/62161/Prospecto
55. Ficha técnica de celebrex 200 mg [Internet]. Agencia Española del Medicamento; abril de 2021. Disponible en: https://cima.aemps.es/cima/dochtml/p/63073/P_63073.html
56. Farré M, Abanades S, Álvarez Y, Barral D, Roset PN. Paracetamol. Dolor. 2004;19(1):5-15.
57. Ficha técnica de paracetamol cinfa [Internet]. Agencia Española del Medicamento; mayo de 2022. Disponible en: https://cima.aemps.es/cima/dochtml/p/70310/P_70310.html
58. Ficha técnica de ibuprofeno 600 mg cinfa [Internet]. Agencia Española del Medicamento, fecha de revisión; marzo de 2023. Disponible en: https://cima.aemps.es/cima/dochtml/p/70039/P_70039.html
59. Ficha técnica de flogoter 25 mg [Internet]. Agencia Española del Medicamento; febrero de 2021. Disponible en: https://cima.aemps.es/cima/dochtml/p/45271/P_45271.html